
Les rhabdomyosarcomes (RMS) sont les sarcomes des tissus mous les plus fréquents chez l’enfant. Ils représentent environ 60% des sarcomes des tissus mous de l’enfant. Néanmoins, comme tous les cancers de l’enfant, c’est une maladie rare.
Ce sont des tumeurs malignes qui affectent principalement les garçons. Au microscope, elles ressemblent aux muscles striés qui permettent de bouger, d’où leur nom grec “cancer des muscles striés”. Elles touchent environ :
- 4 enfants de moins de 15 ans par million/an en France,
- environ 100 enfants/an en France.
Existe-t-il plusieurs types de rhabdomyosarcomes ?
Il existe trois sous-types de rhabdomyosarcomes :
- Les Rhabdomyosarcomes embryonnaires (70 à 80%)
- Les Rhabdomyosarcomes alvéolaires (15 à 20%)
- Les Rhabdomyosarcomes pléiomorphes (plus rares mais plus fréquents chez l’adulte)
A quel âge le rhabdomyosarcome peut-il survenir ?
L’ âge moyen d’un patient atteint de rhabdomyosarcome est de 5 ans, mais ces tumeurs peuvent survenir à tout âge.
On observe cependant deux périodes de pics diagnostiques :
- 2-5 ans
- 15-19 ans
Si ces tumeurs sont majoritairement pédiatriques, elles peuvent aussi être observées chez le jeune adulte, même si cela reste plus rare.
Sur quelles parties du corps les rhabdomyosarcomes se développent-ils ?
Les rhadmyosarcomes peuvent survenir sur n’importe quelle partie du corps :
- La tête et le cou principalement (> 40%)
- Dans les orbites (< 10%)
- L’appareil urogénital (> 30%)
- Sur les membres (10%)
- Ou au niveau du tronc (>15 %)
Les rhabdomyosarcomes présentent un risque de métastase vers les ganglions et les organes de voisinage, mais la forme métastatique d’emblée reste rare (10%).
Quels sont les premiers signes de développement d’un rhabdomyosarcome ?
Les signes évocateurs d’un rhabdomyosarcome dépendent de la localisation initiale et de l’atteinte des organes voisins.
On observe, le plus souvent, les phénomènes suivants :
- Apparition d’une masse ou d’une boule palpable au niveau d’un orifice (vaginal, nasal), d’un membre ou du tronc.
- Rétention aigüe d’urine, difficulté à uriner (pour les RMS urogénitaux)
- Maux de tête, extrusion de l’oeil hors de son orbite, obstruction des sinus.
- Douleurs osseuses
Comment être certain du diagnostic de rhabdomyosarcome ?
La présence des symptômes précédemment évoqués doit conduire à consulter pour entamer un parcours diagnostic afin :
- d’éliminer d’autres diagnostics possibles (tumeurs bénignes, lésions vasculaires ou malformations)
- de poser le diagnostic de rhabdomyosarcome.
Dans un premier temps, le parcours diagnostic doit obligatoirement consister, dans l’ordre, à la réalisation :
- d’examens d’imagerie (Échographie et IRM)
- d’une biopsie (prélèvement de tissus tumoraux par un radiologue ou un chirurgien)
- d’une analyse des tissus (pour déterminer le type exact de tumeur)
- d’une analyse moléculaire (lorsque l’analyse cellulaire n’a pas permis une indentification précise du type de tumeur)
Le caractère malin est parfois difficile à confirmer, notamment chez le nouveau-né ou le nourrisson. Sans confirmation du diagnostic, les tumeurs sont alors classées de malignité “intermédiaire” et d’évolution “incertaine”. C’est pourquoi, afin de pouvoir réaliser des analyses biologiques complémentaires, tout prélèvement d’une masse chez l’enfant doit s’accompagner, au diagnostic, d’une congélation d’une partie de la tumeur.
Dans un second temps, il s’orientera vers la réalisation d’un bilan dit “d’extension” visant à s’assurer que la maladie ne s’est pas étendue à d’autres parties du corps. Ce bilan comprend principalement :
- Une IRM (de la tumeur et des aires ganglionnaires de drainage)
- Un scanner (des zones osseuses voisines)
- Un scanner pulmonaire
- Une scintigaphie osseuse ou plus fréquemment un TEP Scan
- Un bilan médullaire (ponction et biopsie de moelle osseuse)
Ce bilan d’extension pourra, au besoin, être complété par d’autres examens en présence de situations particulières telles que : localisations paraméningées, épanchements pleuraux, atteintes ganglionnaires, etc…
En quoi consiste le traitement des rhabdomyosarcomes ?
Chaque type de rhabdomyosarcome a une valeur pronostique différente qui nécessite donc la mise en place d’un protocole thérapeutique spécifique, préalablement discuté par une équipe pluridisciplinaire, experte dans le traitement des cancers de l’enfant et membre de la Société Française de lutte contre les Cancers et les leucémies de l’Enfant et de l’adolescent (SFCE).
La prise en charge thérapeutique des rhabdomyosarcomes dépend de plusieurs facteurs qui permettent d’adapter les traitements à l’importance de la maladie :
- la localisation de la tumeur
- ses caractéristiques moléculaires
- sa taille
- son étendue
- l’âge de l’enfant
Elle s’organise généralement comme suit :
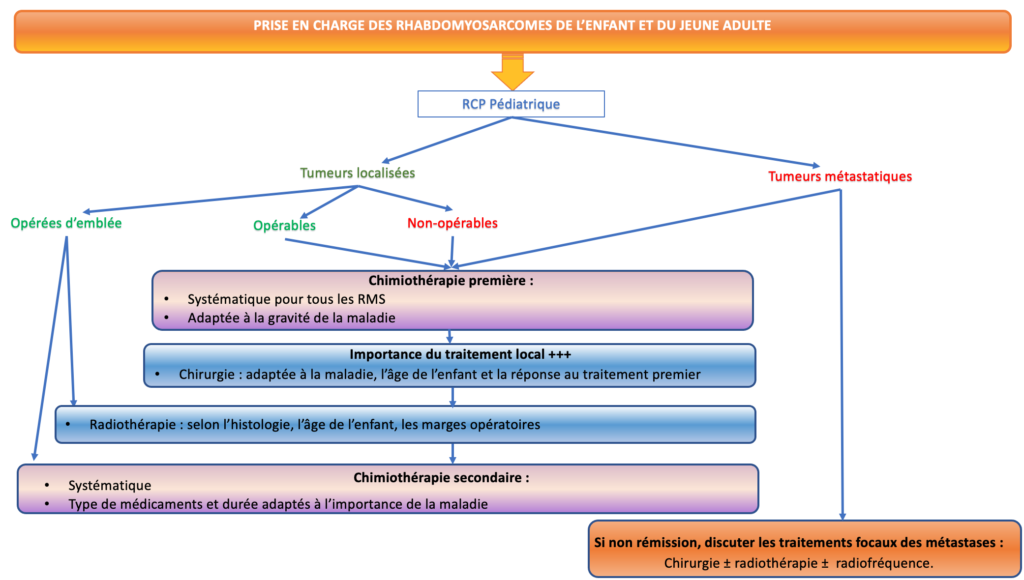
Ces traitements doivent idéalement être initiés dans le cadre d’un protocole de soins européen, ce qui permet de proposer à chaque enfant le meilleur traitement disponible.
Que se passe-t-il à la fin du traitement ?
La fin du traitement marque le début de la période de rémission mais aussi le début de la période de surveillance .
La période de surveillance consiste en la mise en place de différents examens visant à :
- confirmer le maintien de l’état de rémission,
- détecter d’éventuels signes de reprise de la maladie.
A ce jour, la surveillance des rhabdomyosarcomes n’est pas encore codifiée.
Elle pourra néanmoins s’appuyer sur des examens cliniques, des radiographies pulmonaires et des examens d’imagerie du siège de la tumeur primitive qui seront réalisés sur une durée et à une fréquence qui auront préalablement été établies par les médecins.
En cas de reprise de la maladie, la situation du malade devra de nouveau être discutée par l’équipe pluridisciplinaire qui définira la stratégie thérapeutique à mettre en place.
Remerciements : Dr Daniel Orbach, Institut Curie (Paris)